前言:6月19日,由CCHRPP牵头、RDPAC和太美医疗科技共同参与组建的联合专家工作组起草并发布了《临床试验安全性报告工作指引(试行版)》,随后引发了近万名行业同仁的关注与讨论。为了帮助广大的机构和伦理老师在工作中更具体地参考和应用该指引,CCHRPP再度与RDPAC、太美医疗科技携手合作,共同组织专家工作组起草本次SOP模板,并发布如下。
共识专家团队:
白 楠 中国人民解放军总医院
曹 玉 青岛大学附属医院
陈晓云 上海中医药大学附属龙华医院
陈勇川第三军医大学西南医院
樊兴芳 拜耳医药保健有限公司
范侨育 RDPAC PV工作组/辉瑞制药
蒋发烨 广东省人民医院
吉 萍 深圳市生物医学伦理审查委员会
江一峰 上海市第一人民医院
刘海涛 瑞士卫森医药咨询
陆 麒 上海交大医学院附属仁济医院
沈一峰 上海市精神卫生中心
盛艾娟 首都医科大学附属北京佑安医院
沙莉莉 南京鼓楼医院
唐 雪 RDPAC PV工作组/辉瑞制药
万帮喜 太美医疗科技
王美霞 首都医科大学附属北京佑安医院
王 静 RDPAC PV工作组/拜耳医药保健
汪秀琴 江苏省人民医院
吴 蕾 RDPAC
熊宁宁 南京中医药大学附属医院
夏郁松 RDPAC PV工作组/诺华制药
许重远 南方医科大学南方医院
岳 淼 拜耳医药保健有限公司
说明:1.本SOP模板由CCHRPP提供,供各临床试验机构及伦理委员会参考;2.本SOP模板中包含多种流程选择,在有多种流程选择时以【】进行区分;其它的流程选择在{ }中列出;3.对SOP模板的使用说明,以斜体字在{ }中列出;4.本模板中,向临床试验机构及伦理递交SUSAR报告的流程,以申办者向研究者报告,研究者审阅后向临床试验机构及伦理递交。
临床试验机构SAE/SUSAR报告管理流程
版本:
日期:2020-7-20
说明:
本SOP模板由CCHRPP提供,供各临床试验机构及伦理委员会参考;
本SOP模板中包含多种流程选择,在有多种流程选择时以【】进行区分;其它的流程选择在{ }中列出;
对SOP模板的使用说明,以斜体字在{ }中列出;
本模板中,向临床试验机构及伦理递交SUSAR报告的流程,以申办者向研究者报告,研究者审阅后向临床试验机构及伦理递交。
1.目的
本文件为满足2020版《药物临床试验质量管理规范》及其他药物临床试验相关法律法规、指南文件的要求而制定。
本文件规范了本中心开展临床试验时安全性信息的收集、填写、报告,及安全性报告签收阅读、审查和提供回执的整体管理流程。
2.范围
本文件适用于药企申办发起的,用于注册目的的药物临床试验。
本文件适用于研究者、申办者临床试验相关人员(如CRC、CRA)、临床试验机构(称作本机构)和伦理委员会人员(称作本伦理委员会)。
本文件中安全性报告包括个例SAE报告、SUSAR报告及DSUR报告。
本文件适用于指导研究者填写SAE报告至申办者,以及研究者、本机构、本伦理委员会进行SUSAR报告的签收、审阅及审查。
本文件不包括为处理SAE相关的调查、保险理赔等工作内容。
本文件不适用于器械相关的临床试验。
术语与定义
SAE(严重不良事件,Serious Adverse Event,SAE),指满足以下情形中一条或多条的不良事件:
导致死亡;
危及生命,指严重病人即刻存在死亡的风险,并非是指假设将来发展严重时可能出现死亡;
导致住院或住院时间延长;
永久或显著的功能丧失;
致畸、致出生缺陷;
其他重要医学事件:必须运用医学和科学的判断决定是否为重要医学事件,可能不会立即危及生命、死亡或住院,但需要采取医学措施来预防如上情形之一的发生。
SUSAR(可疑非预期严重不良反应,Suspected Unexpected Serious Adverse Reaction,SUSAR):所有与试验药物或上市后药品肯定相关或可疑的非预期且严重的不良反应。在一份SAE报告中如有多个事件,如其中一个事件满足SUSAR标准,则该份报告被评价为SUSAR报告。
DSUR(研发期间安全性更新报告,Development Safety Updated Report,DSUR):依照ICH要求对研发中的药物(包括已批准但仍在进一步研究的药物)进行安全评估的定期报告的通用标准文件。
报告SAE:指的是研究者在获知SAE后的一定时限内将SAE的相关信息传递至申办者的过程。
分发SUSAR:指的是申办者对SAE处理评估后,将判断为SUSAR的报告递交至所有参与同一个药物临床试验的研究者的过程。
签收阅读:指的是研究者收到来自申办者的安全性报告进行签收和审阅,并生成回执传输至申办者。
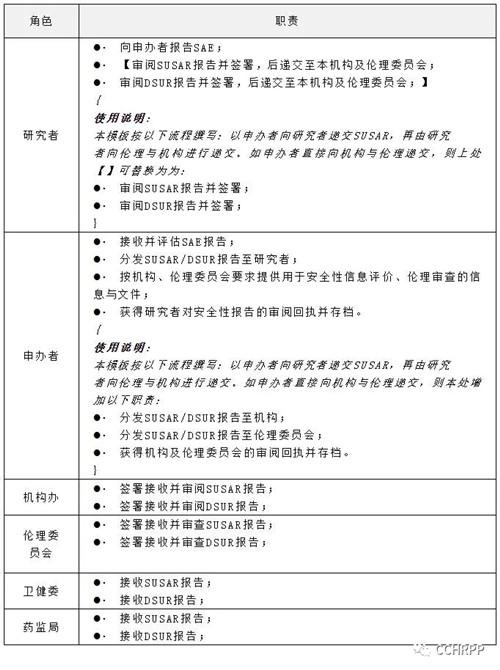
4.职责

5.流程
5.1流程图

使用说明:
本模板按以下流程撰写:以申办者向研究者递交SUSAR,再由研究者向伦理与机构进行
递交。如申办者直接向机构与伦理递交,需去掉上图中的蓝色线条,并将虚线改为实线。
5.2流程说明
5.2.1 SAE报告流程
在本中心开展的临床试验,研究者在获知SAE后,使用标准化SAE表格,立即向申办者书面报告。
申办者按内部标准操作流程进行综合分析评估,判断接收到的SAE报告是否为可疑非预期严重不良反应(SUSAR)。
5.2.2 SUSAR报告流程
申办者将判断为可疑非预期严重不良反应(SUSAR),不论是否为本中心发生,只要涉及的研究药物在本中心开展临床研究,均应快速报告给本中心研究者、临床试验机构和伦理委员会,同时上报国家卫健委、药监局。申办者可使用标准化SUSAR表格进行报告。
伦理委员会针对收到的每一份SUSAR报告给出审查意见及建议。
【同时伦理委员会药物安全专员应负责进行SUSAR报告的登记。】
5.2.3 DSUR报告流程
申办者定期向研究者、本机构、本伦理委员会以及药品监督管理局药品审评中心(CDE)提交DSUR报告。申办者使用DSUR的执行概要进行递交。伦理委员会在收到DSUR后将进行审核。
6.报告要求
6.1概述
本文件中涉及的安全性报告类型、报告途径及时限等要求总结如下:

{使用说明:如果使用信息化系统,且信息化系统可以支持,则按以下要求执行}

6.2流程要求描述
6.2.1 报告填写与确认
按照ICH E2B(R3)标准字段进行SAE信息的填写。
研究者对SAE报告进行审核,确保报告内容完整、准确,进行审阅、进行必要的修改,完成严重性判断及因果评价,以供申办者评估。
6.2.2 报告时限
研究者应在获知SAE的24小时内(除方案另有规定)将报告发送至申办者。
针对SUSAR报告,申办者应当遵循7天和15天的快速报告要求向研究者、临床试验机构及伦理委员会递交。即:
(1) 对于致死或危及生命的SUSAR,申办者应在首次获知后7天内递交,并在随后的8天内报告、完善随访信息(申办者首次获悉当天为第0天);
(2) 对于非致死或危及生命的非预期严重不良反应(SUSAR),申办者应在首次获知后15天内递交。
DSUR报告按年递交,时限与向CDE递交DSUR相同:数据锁定点后60天内。
6.2.3 回执确认
所有递交动作均应有回执:
(1) 申办者收到研究者报告的SAE后,向研究者发送收到回执;
(2) 申办者向研究者、本机构、本伦理中心分发SUSAR报告后,均应获得相应的回执,以作为有效送达之证明。
【接收者通过邮件进行回复,确认已收到,并审阅该安全性文件。邮件的自动回复,不可以作为有效回执。】
{当使用信息化系统时,可以使用电子签名作为查阅的回执。【通过信息化系统进行安全性报告的分发,所有接收者按照系统操作指引进行审阅并电子签名。电子签名内容包括:手写签名图片+签名时间+此签名的含义】}
6.2.4盲态保持
申办者在向本机构、伦理委员会及研究者递交资料时务必应保证信息的盲态(研究者破盲除外)。
6.2.5 安全性信息分析
【本机构及伦理委员会药物安全专员将查阅整理的安全性信息Excel表了解SAE发生的总体情况,并判断是否有预警信息。】
{使用说明:当使用信息化系统且信息化系统功能可支持统计分析时,通过系统中的分析统计功能关注潜在风险【本机构及伦理委员会药物安全专员将借助信息化系统功能判断是否有预警信息。】}
必要时将召开伦理会议审查安全性信息。
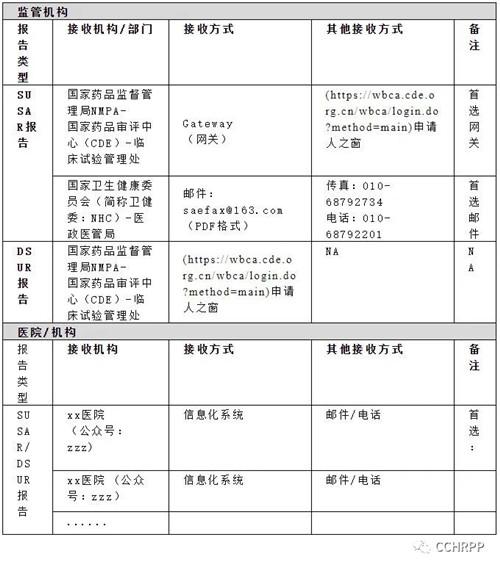
6.2.6安全性报告接收路径
7.参考文件
1) 《药物临床试验质量管理规范》,2020,NMPA及卫健委
2) ICH E6 (R2)Good Clinical Practice,2016,ICH
3) ICH 《E2B(R2)安全性消息处理和个例安全性报告技术规范》,2018, 国家药品监督管理局
4) 《药物临床试验期间安全性数据快速报告标准和程序》,2018,国家药品监督管理局
5) 药品审评中心发布《关于药物临床试验期间安全性数据快速报告标准和程序》有关事项的通知, 2018, 国家药品监督管理局药品审评中心
6) ICH E2F Development safety update report,2011,ICH
7) Guidance for Clinical Investigators, Sponsors, and IRBs. Adverse Event Reporting to IRBs — Improving Human Subject Protection, 2009, FDA
8) 临床试验安全性报告工作指引(试行版),CCHRPP,2020
文章原创来于:C&R专家工作组
文章来自:(搜狐)
(本内容属于网络转载,文中涉及图片等内容如有侵权,请联系编辑删除。市场有风险,选择需谨慎!此文仅供参考,不作买卖及投资依据。)
原创文章,作者:陈晨,如若转载,请注明出处:https://www.kejixun.co/article/501318.html